February is American Heart Month. Heart disease is the leading cause of death for men and women in the United States, causing 1 in 4 deaths each year. We spoke with Biophysical Society members Daniel Beard, University of Michigan, and Andrew McCulloch, University of California, San Diego, about each of their labs’ research related to heart disease.
 Daniel Beard
Daniel Beard
University of Michigan
What is the connection between your research and heart disease? Why is your research important to those concerned about heart disease?
A major focus of our research is on the link between cardiac function and myocardial metabolism. In physical exercise, as the rate of work done by the heart increases compared to rest, the rate of cardiac ATP hydrolysis increases commensurately. By building computer models to represent kinetic control of oxidative (mitochondrial) ATP synthesis, we have been able to build a robust framework for simulating ATP supply/demand matching in vivo [1, 2]. Once we had a working understanding of physiological regulation of ATP synthesis in the heart, we used that working knowledge as a starting place to explore how the physiological system becomes dysfunction in heart disease. In particular, it has long been recognized that in heart failure—a condition in which the mechanical pumping ability of the heart is compromised—concentrations of adenosine triphosphate (ATP), its hydrolysis products, and related metabolites are depleted compared to normal. Neither the causes nor the consequences of these changes are well understood. We think that one consequence of impaired energy metabolism is that it directly contributes to impaired mechanical function. Our current research is squarely focused on determining these causes and consequences of mechano-energetic dysfunction, and on finding new ways to repair/restore myocardial metabolism to improve mechanical function in heart failure.
How did you get into this area of research?
My interest in this area goes back to work that was part of my PhD thesis, working in Jim Bassingthwaighte’s lab on simulating physiological transport of oxygen, and other solutes. I became increasingly interested in oxygen transport, and questions of how oxygen delivery is matched to oxygen demand in the myocardium. In this context I started to look in some depth at the physiological regulation of oxidative phosphorylation, not for its own sake, but because I was interested in how it contributed to governing oxygen transport. In other words, at first mitochondrial metabolism was on the periphery of what I was initially interested in. That peripheral interest gradually grew into a major research thrust.
Do you receive public funding for this work? If so, from what agency?
Yes, we have been fortunate to be supported for this work, primarily from the National Institutes of Health National Heart Lung and Blood Institute.
Have you had any surprise findings thus far?
Our first big surprise followed from my lab’s first NIH funding award, for which we proposed to investigate a conundrum in the field cardiac energy metabolism: How is ATP synthesis supply matched to changing demand levels in light of apparently constant concentrations of ADP and inorganic phosphate (Pi) in the heart? Our answer to this question is that, in fact, the ATP supply is not matched to demand while maintaining constant levels of ADP and Pi. Specifically, our model-based analysis of the in vivo data on phosphate metabolite levels in the heart predicts that changing [Pi] provides a critical feedback signal to stimulate increasing ATP synthesis with increasing rates of ATP hydrolysis. This simple idea fundamentally challenges established ideas in the field. In fact, our hypothesized role of Pi as a primary feedback signal for oxidative ATP synthesis in the heart is still at least a little bit controversial and it would be incorrect to say that it is universally accepted. Regardless, the hypothesis has survived all of our attempts to disprove it!
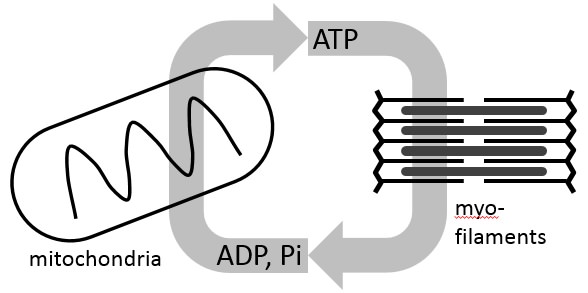
he next big surprise came when we applied our models of cardiac energy metabolism to analyze data from animal models of heart failure. We found that hallmarks of myocardial energetics in heart failure—diminished ATP and ATP hydrolysis potential—could be effectively captured by simulations in which mitochondrial function is normal. At first blush that finding seems contradictory: Since 95% of ATP in the heart is produced by mitochondria, how can diseased hearts with diminished ATP have normally functioning mitochondria? The explanation pointed to by our analysis is that reduction in cytoplasmic metabolic pools is a critical driver of energetic/metabolic dysfunction in the failing heart. Those predictions were later put to the test when, in collaboration with Igor Efimov’s lab, we were able to measure mitochondrial function in samples from failing human hearts, revealing no significant dysfunction compared to healthy controls [3]. This is not to say that mitochondrial function is normal in the heart in heart failure, but rather that we believe dysfunction in mitochondrial energy metabolism is not necessarily intrinsic to the mitochondria themselves, but rather driven by the local environment they find themselves in.
What is particularly interesting about the work from the perspective of the public?
Patients with heart failure wind up seeing a cardiologist, not with complaints of impaired cardiac energetics, but with health problems—shortness of breath, exercise intolerance—directly related to impaired heart pumping. Yet we think that metabolic/energetic dysfunction can contribute directly to mechanical dysfunction in heart failure. Our current research in this area is on the link between mechanics and energetics. We are using computer models that integrate metabolic and mechanical function to better understand the physiological connection between chemical and mechanical function in the heart, to determine ways in which this connection breaks down in heart disease, and identify new strategies to improve mechanical pump function by restoring the metabolic state.
- Wu, F., et al., Phosphate metabolite concentrations and ATP hydrolysis potential in normal and ischaemic hearts. J Physiol, 2008. 586(17): p. 4193-208.
- Bazil, J.N., D.A. Beard, and K.C. Vinnakota, Catalytic Coupling of Oxidative Phosphorylation, ATP Demand, and Reactive Oxygen Species Generation. Biophys J, 2016. 110(4): p. 962-71.
- Holzem, K.M., et al., Mitochondrial structure and function are not different between nonfailing donor and end-stage failing human hearts. FASEB J, 2016.
 Andrew McCulloch
Andrew McCulloch
University of California, San Diego
What is the connection between your research and heart disease?
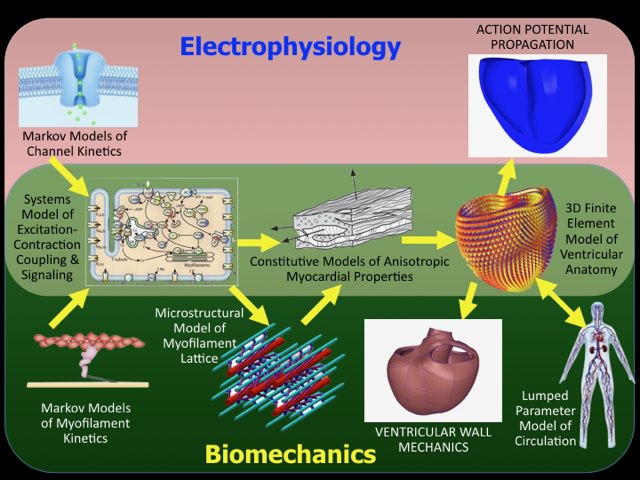
We use in-vitro and in-vivo experiments primarily using mouse models of heart failure and arrhythmias together with multi-scale computational models to discover cellular and molecular mechanisms of electrical and mechanical dysfunction at the tissue and organ scales.
Why is your research important to those concerned about heart disease?
Our research is becoming of particular interest to cardiologists and patients with heart diseases because we have started to discover ways to apply the computational modeling tools we validated in the lab to analyze and predict therapeutic outcomes in patients, including heart failure patients with electrical dyssynchrony who are indicated for cardiac resynchronization therapy, patients with atrial fibrillation or at risk of ventricular fibrillation who can benefit from radiofrequency ablation therapy, and children and adults with congenital heart diseases who are at risk of developing heart failure or arrhythmias later in life.
How did you get into this area of research?
It started as a MS thesis project in Engineering Science when I was 19, that turned into a PhD in Physiology and Engineering and then a faculty career.
How long have you been working on it?
Over 35 years.
Do you receive public funding for this work? If so, from what agency?
Yes, we rely almost exclusively from NIH funding via the NHLBI, NIBIB and NIGMS though we have also received valuable support in the past from the NSF, NASA, DARPA, AHA and Heart Rhythm Society.
Have you had any surprise findings thus far?
Yes, we have found with and novel mouse and computational models that phosphorylation of specific serine residues on cardiac myosin regulatory light chain not only affect crossbridge dynamics, but also give rise to a feedback that affects the calcium-dependent active of the thin filament. We have also found that patient-specific models have the unexpected potential to predict and optimize the outcomes of device therapies for heart failure.
What is particularly interesting about the work from the perspective of other researchers?
The power of modern multi-scale and systems biology modeling to help understand genotype-phenotype relations in animal models of heart diseases.
What is particularly interesting about the work from the perspective of the public?
The potential for computer models to improve the early diagnosis and clinical management of heart diseases in adults and children.